The Regulation (EU) 2024/1860 amending Regulations (EU) 2017/745 (MDR) and (EU) 2017/746 (IVDR) has been published in the Official Journal of the European Union (OJEU). The aim of this legislation is to ensure continuity of supply on the European market for in vitro diagnostic devices. It also amends the provisions relating to the compulsory use of EUDAMED for devices that comply with one of the regulations or were placed on the market under one of the directives (legacy devices). Lastly, the text introduces the obligation for manufacturers and all economic operators in the supply chain to declare supply disruptions or risks of supply disruptions.
Here is the summary of the provisions introduced:
Change in the IVDR transitional provisions
Modification of the calendar
The change of transitional provisions only applies to devices requiring the involvement of a notified body for their conformity assessment under IVDR. The dates after which devices may continue to be placed on the market have been extended by two years, provided that the manufacturer complies with certain conditions:
The manufacturer must:
- Have implemented and maintain a Quality Management System that complies with the requirements of Article 10, paragraph 8 of the IVDR before 26/05/2025.
- Have lodged a formal application for certification for each device with a Notified Body designated against the IVDR the date applicable to the risk class of the device; and
- Have signed a contract with the latter which covers the concerned devices before the date applicable to the risk class of the device.
In addition, the devices:
- Must continue to comply with the Directive requirements;
- Must not undergo any significant change in the design or intended purpose;
- Must not present an unacceptable risk to the health or safety of patients, users or other persons, or to other aspects of the protection of public health.
Important: Devices that have been withdrawn from the IVDD certification scope cannot benefit from the new transitional provisions.
The schedule is defined as follows, based on the IVDR risk classification of devices:
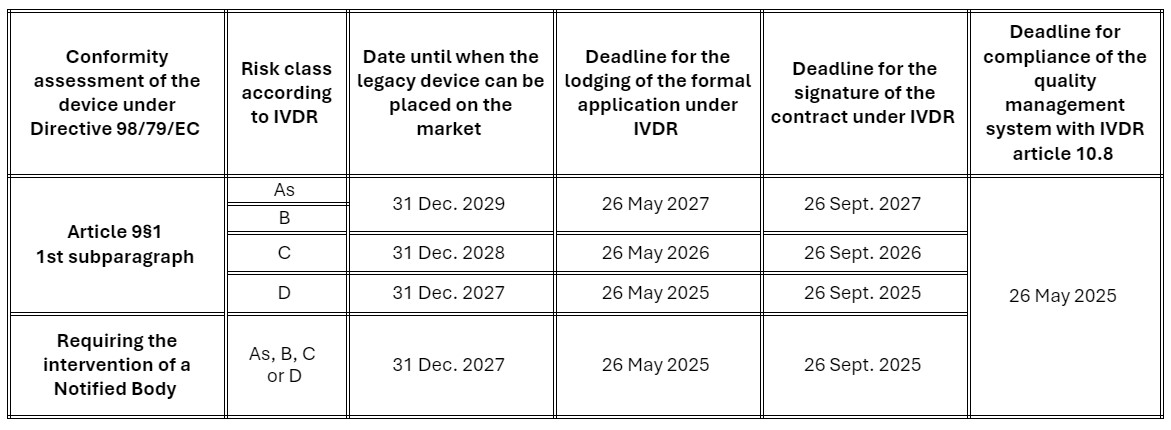
It is important to note that devices that are class A must already comply with all IVDR applicable provisions.
Certificate management
Nothing in the Regulation (EU) 2024/1860 requires the reissuance or modification of IVDD certificates. They are deemed to be de facto extended, provided only that they have not been withdrawn.
Expired certificates covering devices that are in the scope of an IVDR certification contract (written agreement) before its expiry or covered by an IVDR article 54/92 derogation/communication will also be considered valid.
GMED will issue letter that confirms a formal application has been lodged and a contract has been signed with the manufacturer for the certification under IVDR of devices that were placed on the market pursuant Directive 98/79/EC.
Appropriate surveillance
Devices that are covered by a valid certificate shall continue to be under the appropriate surveillance of notified bodies. Which means that audits pursuant IVDD will continue to be performed until the end of the transition period. The notified bodies will continue to assess changes and reports on incidents.
The Regulation (EU) 2024/1860 provides that the Notified Body conducting the conformity assessment procedure under IVDR shall be responsible for the said surveillance from 26 September 2025 on, at the latest.
Other provisions introduced
Declaration of shortage
The Regulation (EU) 2024/1860 also introduced the obligation for the manufacturer and any economic operator involved in the supply chain to declare stock shortages for so-called “critical” devices. This provision also applies to legacy devices.
Modification of the provisions relating to the use of EUDAMED
After the entry into force of the Regulation (EU) 2024/1860, EUDAMED database will be declared functional module by module.
- Legacy devices or devices compliant with the regulations will have to be registered no later than 12 months after publication of the module functionality notice.
- Certificates will have to be registered by notified bodies no later than 18 months after publication of the module’s functionality notice.
Conclusion
As we have seen, the changes made are aiming to maintain the availability of a wide range of in vitro diagnostic devices on the European market, while ensuring a gradual transition to the new regulatory framework.
However, we would like to reiterate that, despite this postponement, manufacturers must continue their transition efforts in order to avoid the risk of an imbalance between the capacity of notified bodies and the demand for certification at the next deadlines.
As a Notified Body, GMED is committed to working with the in vitro diagnostics industry in the certification of legacy devices. To this end, we invite you to contact GMED’s teams today to find out about the solutions we have implemented in relation to the transitional provisions:
- For Europe enquiries, contact GMED SAS: sales@lne-gmed.com
- For America or International enquiries, contact GMED North America: request@lne-gmed.com
share
Events
Find an answer to the challenges you are facing in one of our upcoming events: trainings, webinars, forums...