
Le Règlement (UE) 2024/1860 amendant les Règlements (UE) 2017/745 (MDR) et (UE) 2017/746 (IVDR) a été publié au Journal officiel de l’Union européenne (JOUE). Il a pour objectif principal d’assurer la continuité de l’approvisionnement sur le marché européen des dispositifs de diagnostic in vitro. Il modifie également les dispositions relatives à l’utilisation obligatoire d’EUDAMED pour les dispositifs conformes à l’un des règlements ou mis sur le marché au titre de l’une des directives (legacy devices). Enfin, le texte introduit l’obligation pour les fabricants et tous les opérateurs économiques de la chaîne d’approvisionnement de déclarer les ruptures d’approvisionnement ou les risques de rupture d’approvisionnement.
Les dispositions introduites sont les suivantes :
Modifications des dispositions transitoires au titre de l’IVDR
Modification du calendrier
La modification des dispositions transitoires ne s’applique qu’aux dispositifs nécessitant l’intervention d’un organisme notifié pour leur évaluation de conformité au titre de l’IVDR. Les dates après lesquelles les dispositifs peuvent continuer à être mis sur le marché ont été prolongées de deux ans, à condition que le fabricant respecte certaines conditions :
Le fabricant doit :
- Avoir mis en œuvre et maintenir un système de gestion de la qualité conforme aux exigences de l’article 10, paragraphe 8 de l’IVDR avant le 26/05/2025.
- Avoir déposé une demande formelle de certification pour chaque dispositif auprès d’un organisme notifié désigné au titre de l’IVDR à la date applicable à la classe de risque du dispositif ; et
- Avoir signé un contrat avec ce dernier qui couvre les dispositifs concernés avant la date applicable à la classe de risque du dispositif.
En plus, les dispositifs :
- doivent continuer d’être conformes aux exigences de la directive ;
- ne doivent pas faire l’objet d’une modification significative de leur conception ou de leur destination ;
- ne doivent pas présenter de risque inacceptable pour la santé ou la sécurité des patients, des utilisateurs ou d’autres personnes, ou pour d’autres aspects de la protection de la santé publique.
Important : Les dispositifs qui ont été retirés du périmètre de certification au titre de la directive 98/79/CE ne peuvent pas bénéficier des nouvelles dispositions transitoires.
Le calendrier est défini comme suit, sur la base de la classification des risques des dispositifs par l’IVDR :
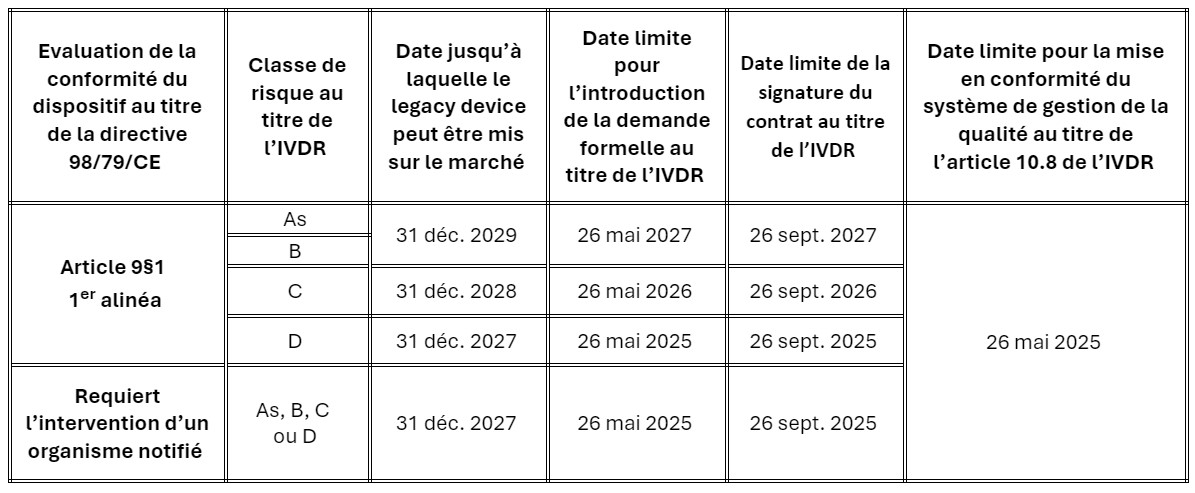
Il est important de noter que les dispositifs de classe A doivent d’ores et déjà être conformes à toutes les dispositions applicables de l’IVDR.
Gestion des certificats
Aucune disposition du Règlement (UE) 2024/1860 n’exige la réémission ou la modification des certificats au titre de la directive 98/79/CE. Ils sont considérés être prolongés de facto, à la seule condition qu’ils n’aient pas été retirés.
Les certificats expirés couvrant des dispositifs qui sont dans le périmètre d’un contrat de certification IVDR (accord écrit) avant son expiration ou couverts par une dérogation/communication émise au titre de l’article 54/92 de l’IVDR seront également considérés comme valides.
GMED émettra une lettre confirmant qu’une demande formelle a été déposée et qu’un contrat a été signé avec le fabricant pour la certification IVDR des dispositifs qui ont été mis sur le marché au titre de la directive 98/79/CE.
Surveillance appropriée
Les dispositifs couverts par un certificat valide continuent à faire l’objet d’une surveillance appropriée par les organismes notifiés. Cela signifie que les audits effectués conformément à la directive sur les dispositifs médicaux in vitro continueront d’être réalisés jusqu’à la fin de la période de transition. Les organismes notifiés continueront à évaluer les changements et les rapports de vigilance.
Le Règlement (UE) 2024/1860 prévoit que l’organisme notifié qui effectue la procédure d’évaluation de la conformité au titre de l’IVDR soit responsable de ladite surveillance à partir du 26 septembre 2025 au plus tard.
Autres dispositions introduites
Déclaration de rupture de stock
Le Règlement (UE) 2024/1860 a également introduit l’obligation pour le fabricant et tout opérateur économique impliqué dans la chaîne d’approvisionnement de déclarer les ruptures de stock pour les dispositifs dits « critiques ». Cette disposition s’applique également aux legacy devices.
Modification des dispositions relatives à l’utilisation d’EUDAMED
Après l’entrée en vigueur du Règlement (UE) 2024/1860, la base de données EUDAMED sera déclarée fonctionnelle module par module.
- Les legacy devices ou dispositifs conformes aux règlements devront être enregistrés au plus tard 12 mois après la publication de l’avis de fonctionnalité du module.
- Les certificats devront être enregistrés par les organismes notifiés au plus tard 18 mois après la publication de l’avis de fonctionnalité du module.
Conclusion
Comme nous avons pu le voir, les modifications apportées visent à maintenir la disponibilité sur le marché européen d’un large éventail de dispositifs de diagnostic in vitro tout en assurant la transition progressive vers le nouveau cadre règlementaire.
Nous tenons cependant à rappeler que, malgré ce report, les fabricants doivent poursuivre leurs efforts de transition afin d’éviter, aux prochaines échéances, le risque d’un déséquilibre entre le capacitaire des organismes notifiés et les demandes de certification.
GMED, en tant qu’Organisme Notifié, s’engage auprès de l’industrie des dispositifs de diagnostic in vitro dans l’effort de certification des legacy devices. Pour ce faire, nous vous invitons à contacter dès à présent les équipes de GMED afin de connaître les solutions mises en œuvre relative aux dispositions transitoires : sales@lne-gmed.com
PARTAGER
Événements
Pour répondre aux enjeux auxquels vous êtes confrontés, prenez connaissance des prochains événements : formations, webinars, forums…